
Lipoidozė (lizosomų lipidų kaupimosi liga): kas tai yra, pavyzdžiai
Dėmesio! Informacija skirta tik informaciniams tikslams ir nėra skirta diagnozuoti ir skirti gydymą. Visada pasitarkite su gydytoju specialistu!
Turinys
- Kas yra lizosomų kaupimosi ligos?
- Kas yra lipidai?
- Kaip paveldima lipoidozė?
- Lizosomų kaupimosi ligų tipai ir simptomai
Kas yra lizosomų kaupimosi ligos?
Lizosomų kaupimosi ligos (lipoidozė), yra paveldimų medžiagų apykaitos ligų grupė, kurios metu įvairiose organizmo ląstelėse ir audiniuose kaupiasi kenksmingi riebalinių medžiagų (lipidų) kiekiai.
Šių sutrikimų turintys žmonės arba negamina pakankamai vieno iš fermentų, reikalingų lipidams skaidyti (metabolizuoti), arba gamina fermentus, kurie neveikia tinkamai.
Laikui bėgant šis per didelis riebalų kaupimasis gali sukelti nuolatinį ląstelių ir audinių pažeidimą, ypač smegenyse, periferinėje nervų sistemoje (nervuose nuo nugaros smegenų iki kitų kūnas), kepenys, blužnis ir kaulų čiulpų.
Kas yra lipidai?
Lipidai yra į riebalus panašios medžiagos, kurios yra svarbios ląstelės viduje ir tarp jų esančios membranos bei mielino apvalkalo dalys, dengiančios ir apsaugančios nervus. Lipidai apima aliejus, riebalų rūgštis, vaškus, steroidus (tokius kaip cholesterolis ir estrogenas) ir kitus susijusius junginius.
Šios riebalinės medžiagos natūraliai kaupiamos organizmo ląstelėse, organuose ir audiniuose. Maži kūnai ląstelėse, vadinami lizosomomis, reguliariai paverčia arba metabolizuoja lipidus ir baltymus į mažesnius komponentus, kad aprūpintų kūną energija. Sutrikimai, kai lizosomose lieka viduląstelinė medžiaga, kurios negalima metabolizuoti, vadinami lizosomų kaupimosi ligomis.
Be ligų, susijusių su lipidų kaupimu, kitos lizosominės ligos, susijusios su lipidų kaupimu, yra mukolipidozės, kurių metu kaupiasi ląstelės ir audiniai. per didelis lipidų kiekis su prie jų prisirišusiomis cukraus molekulėmis, taip pat mukopolisacharidozės, kuriose susikaupia per didelis kiekis didelio sudėtingo cukraus. molekules.
Kaip paveldima lipoidozė?
Lizosomų kaupimosi ligos yra paveldimos iš vieno ar abiejų tėvų, turinčių sugedusį geną, reguliuojantį specifinį lipidus metabolizuojantį fermentą tam tikroje kūno ląstelių klasėje. Jie gali būti paveldimi dviem būdais:
- Autosominis recesyvinis paveldėjimas atsiranda, kai abu tėvai nešioja ir perduoda sugedusio geno kopiją, tačiau nė vienas iš tėvų nėra paveiktas šio sutrikimo. Kiekvienas vaikas, gimęs šiems tėvams, turi 25 procentų tikimybę, kad paveldės abi sugedusio geno kopijas, 50 procentų tikimybė, kad jis bus nešiotojas, panašus į savo tėvus, ir 25 procentų tikimybė, kad nepaveldės jokios sugedusios kopijos. genas. Bet kurios lyties vaikai gali būti paveldimi autosominiu recesyviniu būdu.
- Su X susijęs (arba su lytimi susijęs) recesyvinis paveldėjimas atsiranda, kai motina X chromosomoje nešioja pažeistą geną. X ir Y chromosomos dalyvauja nustatant lytį. Moterys turi dvi X chromosomas, o vyrai turi vieną X chromosomą ir vieną Y chromosomą. Moterų nešiotojų sūnūs turi 50 procentų galimybę paveldėti ir paveikti sutrikimą, nes sūnūs gauna vieną X chromosomą iš savo motinos ir vieną Y chromosomą iš savo tėvo. Dukterys turi 50 procentų galimybę paveldėti pažeistą X chromosomą iš savo motinos ir yra nešiotojos arba nesunkiai paveiktos. Sergantys vyrai šios ligos neperduoda savo sūnums, tačiau jų dukros bus ligos nešiotojai ir nešiotojai.
Lizosomų kaupimosi ligų tipai ir simptomai
Gošė liga - dažniausia iš lizosominių ligų. Šią ligą sukelia fermento gliukocerebrozidazės trūkumas, dėl kurio gliukocerebrozidas kaupiasi daugelyje audinių. Riebalinės medžiagos gali kauptis smegenyse, blužnyje, kepenyse, inkstuose, plaučiuose ir kaulų čiulpuose.
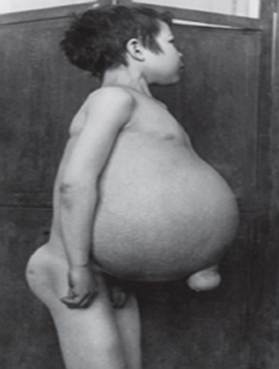
Simptomai gali būti smegenų pažeidimas, blužnies padidėjimas ir kepenys, sutrikusi kepenų funkcija, skeleto sutrikimai ir kaulų pažeidimai, galintys sukelti skausmą ir lūžius, limfmazgių ir (kartais) gretimų sąnarių patinimą, pilvo pūtimas, rusvas odos atspalvis, anemija, mažas trombocitų skaičius ir geltonos dėmės ant akių.
Sunkiausiai paveikti asmenys taip pat gali būti jautresni infekcijoms. Liga vienodai paveikia vyrus ir moteris.
Gošė liga turi tris bendruosius klinikinius potipius:
- 1 tipas (Arba ne neuronopatinis tipas) yra labiausiai paplitusi ligos forma JAV ir Europoje. Smegenys nepažeidžiamos, tačiau gali būti pažeisti plaučiai ir retais atvejais inkstai. Simptomai gali prasidėti ankstyvame amžiuje arba sulaukus pilnametystės, įskaitant kepenų padidėjimą ir stiprų blužnies padidėjimą, kuris gali sukelti plyšimą ir papildomų komplikacijų. Skeleto silpnumas ir kaulų liga gali būti plati. Šios grupės žmonės paprastai lengvai atsiranda mėlynių dėl mažo trombocitų kiekio kraujyje. Jie taip pat gali jausti nuovargį dėl anemijos. Priklausomai nuo ligos pradžios ir sunkumo, žmonės, sergantys 1 tipas gali gyventi iki pilnametystės. Daugelis sergančiųjų serga lengva liga arba gali nepasireikšti jokių simptomų. nors 1tipo Gošė liga yra paplitusi tarp aškenazių žydų paveldo žmonių ir gali paveikti visų etninių grupių žmones.
- 2 tipas (arba ūminė neuropatija vaikystėje Gošė liga) paprastai prasideda per 3 mėnesius nuo gimimo. Simptomai yra platus ir progresuojantis smegenų pažeidimas, spazmiškumas, traukuliai, galūnių sustingimas, kepenų padidėjimas.kepenų hepatomegalija) ir blužnis (splenomegalija), nenormalūs akių judesiai ir silpnas gebėjimas čiulpti bei nuryti. Sergantys vaikai paprastai miršta nesulaukę 2 metų.
- 3 tipas (lėtinis neuronopatinis forma) gali prasidėti bet kada vaikystėje ar net suaugus. Jai būdingi lėtai progresuojantys, bet ne tokie sunkūs neurologiniai simptomai, lyginant su ūmine Gošė liga 2 tipai. Pagrindiniai simptomai yra akių judėjimo sutrikimai, pažinimo sutrikimai, bloga koordinacija, traukuliai, blužnies ir (arba) kepenų padidėjimas, skeleto sutrikimai, kraujo sutrikimai, įskaitant anemiją, ir kvėpavimas. Beveik visi, sergantys Gošė liga 3 tipai, kuriems bus taikoma pakaitinė fermentų terapija, sulauks pilnametystės.
Pacientams 1 ir 3 tipai Pakaitinė fermentų terapija, švirkščiama į veną kas dvi savaites, gali žymiai sumažinti kepenų ir blužnies dydį, sumažinti skeleto anomalijas ir panaikinti kitas apraiškas. Sėkminga kaulų čiulpų transplantacija išgydo neurologines ligos apraiškas. Tačiau ši procedūra kelia didelę riziką ir retai atliekama Gošė liga sergantiems asmenims.
Retais atvejais gali prireikti operacijos, kad būtų pašalinta visa blužnis arba jos dalis (jei žmogaus trombocitų skaičius labai mažas arba kai padidėjęs organas labai paveikia asmens komfortą). Kai kuriems anemija sergantiems žmonėms gali būti naudingas kraujo perpylimas. Kitiems gali prireikti sąnario pakeitimo operacijos, kad pagerėtų mobilumas ir gyvenimo kokybė. Šiuo metu nėra veiksmingo smegenų pažeidimo, kuris gali atsirasti žmonėms, sergantiems 2 ir 3 tipo Gošė liga, gydymo.
Niemann-Pick liga yra autosominių recesyvinių sutrikimų grupė, kurią sukelia riebalų ir cholesterolio kaupimasis kepenų, blužnies, kaulų čiulpų, plaučių ir kai kuriais atvejais smegenų ląstelėse.
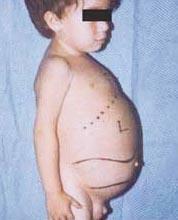
Neurologinės komplikacijos gali būti ataksija (raumenų koordinacijos stoka, galinti turėti įtakos vaikščiojimui, rašymui ir valgymui, be kitų funkcijų), akių paralyžius, smegenų degeneracija, mokymosi problemos, spazmiškumas, pasunkėjęs maitinimas ir rijimas, neryškus kalba, raumenų tonuso praradimas, padidėjęs jautrumas lytėjimui ir tam tikras ragenos drumstumas dėl per didelio susikaupimo medžiagų. 50% paveiktų žmonių aplink tinklainės centrą susidaro būdinga vyšnių raudonumo aureolė, kurią gydytojas gali pamatyti specialiu instrumentu.
Niemann-Pick liga skirstoma į tris bendrus potipius:
- A tipas, sunkiausia forma, prasideda ankstyvoje vaikystėje. Gimę kūdikiai atrodo normalūs, tačiau iki 6 mėnesių išsivysto gilus smegenų pažeidimas, padidėja kepenys ir blužnis, padidėja limfmazgiai ir poodiniai mazgai (ksantomos). Blužnis gali tapti 10 kartų didesnė už įprastą dydį ir gali plyšti, sukeldama kraujavimą. Šie vaikai laipsniškai silpnėja, praranda motorinę funkciją, gali susirgti anemija ir yra linkę į pasikartojančias infekcijas. Jie retai gyvena ilgiau nei 18 mėnesių. Ši ligos forma labiausiai paplitusi žydų šeimose.
- B tipas(arba paauglystės pradžia) dažniausiai nepaveikia smegenų, tačiau dauguma vaikų vystosi ataksija, nervų, paliekančių nugaros smegenis, pažeidimas (periferinė neuropatija) ir su amžiumi progresuojantys plaučių sutrikimai. Paaugliams būdingas kepenų ir blužnies padidėjimas. Žmonės su B tipas gali gyventi gana ilgai, tačiau daugeliui jų reikia visą gyvenimą trunkančios deguonies terapijos dėl plaučių pažeidimo. Niemann-Pick A ir B tipai yra riebalinės medžiagos, vadinamos sfingomielinu, kaupimosi dėl fermento, vadinamo sfingomielinaze, trūkumo.
- C tipas gali atsirasti ankstyvame gyvenime arba išsivystyti paauglystėje ar net pilnametystėje. Niemann-Pick ligaC tipas sukelia ne sflingomielinazės trūkumas, o NPC1 ar NPC2 baltymų trūkumas. Dėl to įvairūs lipidai ir ypač cholesterolis kaupiasi nervinėse ląstelėse ir sukelia jų veiklos sutrikimus. Smegenys gali būti plačiai paveiktos, dėl to negalėjimas žiūrėti aukštyn ir žemyn, sunku vaikščioti ir ryti, progresuoja klausos praradimas ir progresuoja. demencija. Žmonės su C tipas yra tik nedidelis blužnies ir kepenų padidėjimas. Žmonių, sergančių C tipas labai skiriasi. Kai kurie žmonės miršta vaikystėje, o kiti, kurie yra mažiau paveikti, taip pat gali gyventi suaugę.
Šiuo metu Niemann-Pick liga nėra išgydoma. Gydymas yra palaikomasis. Vaikai dažniausiai miršta nuo infekcijos ar progresuojančio neurologinio netekimo. Buvo kaulų čiulpų transplantacijos atvejų keliems pacientams, sergantiems B tipas su įvairiais rezultatais.
Fabry ligataip pat žinomas kaip alfa-galaktozidazės-A trūkumas, sukelia riebalinių medžiagų kaupimąsi autonominėje nervų sistemoje (toje nervų sistemos dalyje, kuri kontroliuoja nevalingas funkcijas, tokias kaip kvėpavimas ir širdies plakimas), akių, inkstų ir širdies ir kraujagyslių sistemos sistema.
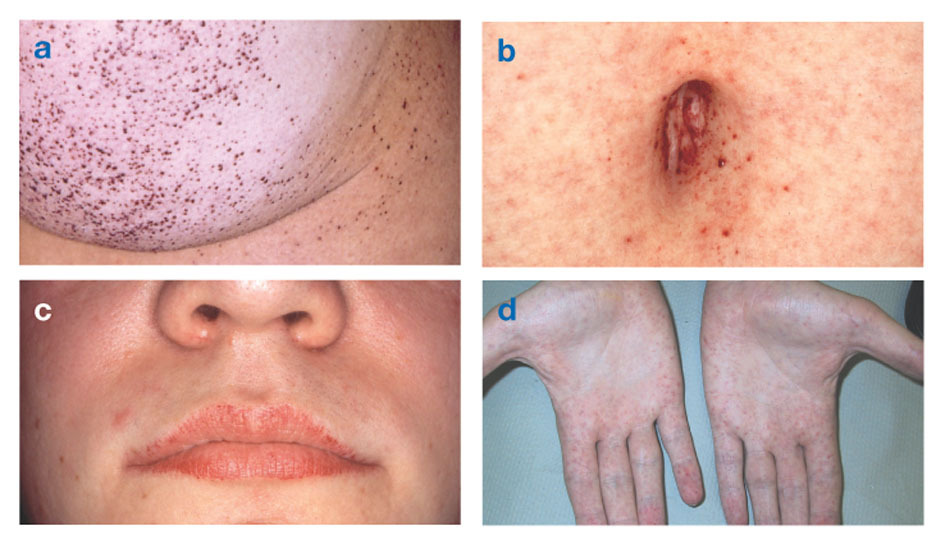
Fabry liga yra vienintelė su X susijusi lipidų liga. Šia liga daugiausia serga vyrai, nors moterims pasireiškia lengvesnė ir įvairesnė forma. Retai paveiktoms moterims būdingi sunkūs simptomai, panašūs į tuos, kurie pasireiškia šia liga sergantiems vyrams. Simptomai dažniausiai pasireiškia vaikystėje ar paauglystėje.
Neurologiniai požymiai yra deginantis rankų ir kojų skausmas, kuris sustiprėja karštu oru arba po jo pratimai, taip pat medžiagos perteklius permatomuose ragenos sluoksniuose (dėl to drumsčiasi, bet be regėjimo pokyčiai). Riebalų kaupimasis kraujagyslių sienelėse gali sutrikdyti kraujotaką, todėl žmogui kyla pavojus insultas arba širdies smūgis. Kiti simptomai yra padidėjusi širdis, progresuojanti inkstų nepakankamumassukeliančios galutinės stadijos inkstų ligą, virškinimo trakto sutrikimus, sumažėjusį prakaitavimą ir karščiavimą.
Angiokeratomos (mažos, ne vėžinės, rausvai violetinės iškilusios odos dėmės) gali išsivystyti apatinėje liemens dalyje ir dažniau su amžiumi.
Žmonės, sergantys Fabry liga, dažnai miršta per anksti nuo komplikacijų širdies nepakankamumas, inkstų nepakankamumas arba insultas. Vaistai, tokie kaip fenitoinas ir karbamazepinas, dažnai skiriami skausmui, kurį lydi Fabry liga, gydyti, bet ne jam gydyti. Metoklopramidas arba Lipisorbas (maisto papildas) gali sumažinti virškinimo trakto sutrikimus dažnai pasireiškia žmonėms, sergantiems Fabry liga, o kai kuriems žmonėms gali prireikti persodinti inkstą arba dializė. Kai kuriems Fabry liga sergantiems žmonėms fermentų pakeitimas gali sumažinti saugojimą, sumažinti skausmą ir išsaugoti organų funkciją.
Farberio liga taip pat žinomas kaip Farberio lipogranulomatozė, aprašoma grupė retų autosominių recesyvinių sutrikimų, sukeliančių riebalinių medžiagų kaupimąsi sąnariuose, audiniuose ir centrinėje nervų sistemoje. Tai paveikia ir vyrus, ir moteris. Liga dažniausiai prasideda ankstyvoje vaikystėje, tačiau gali pasireikšti ir vėliau.
Vaikams, sergantiems klasikine Farberio liga, išsivysto neurologiniai simptomai, kurie gali apimti padidėjusį mieguistumą ir vangumą bei su rijimas. Taip pat gali būti pažeistos kepenys, širdis ir inkstai. Kiti simptomai gali būti sąnarių kontraktūros (lėtinis raumenų ar sausgyslių sutrumpėjimas aplink sąnarius), vėmimas, artritas, padidėjimas. limfmazgiai, sąnarių uždegimas, užkimimas ir gumbeliai po oda, kurie sustorėja aplink sąnarius progresuojant ligų.
Sergantiems žmonėms, kuriems sunku kvėpuoti, gali tekti įkišti kvėpavimo vamzdelį. Dauguma vaikų, sergančių šia liga, miršta iki 2 metų amžiaus, dažniausiai nuo plaučių ligos. Esant vienai sunkiausių ligos formų, netrukus po gimimo galima diagnozuoti kepenų ir blužnies padidėjimą. Kūdikiai, gimę su šia ligos forma, paprastai miršta per 6 mėnesius.
Farberio ligą sukelia fermento, vadinamo keramidaze, trūkumas. Šiuo metu nėra specifinio Farberio ligos gydymo. Skausmui malšinti gali būti skiriami kortikosteroidai. Kaulų čiulpų transplantacija gali sumažinti granulomų (mažų uždegiminių audinių masių) skaičių žmonėms, sergantiems lengvu uždegimu arba pacientams, kuriems nėra plaučių ar nervų sistemos komplikacijų. Vyresnio amžiaus žmonėms granulomos gali būti sumažintos arba pašalintos chirurginiu būdu.
Gangliozidozė susideda iš dviejų skirtingų genetinių ligų grupių. Abi yra autosominės recesyvinės ir vienodai veikia tiek vyrus, tiek moteris.
GM1 gangliozidozė.
GM1 gangliozidozę sukelia fermento beta-galaktozidazės trūkumas, dėl kurio nenormaliai kaupiasi rūgštinės lipidinės medžiagos, ypač centrinės ir periferinės nervų ląstelėse sistemos. GM1 gangliozidozė turi tris klinikinius pasireiškimus:
- GM1 (sunkiausias potipis, atsirandantis netrukus po gimimo) gali apimti neurodegeneraciją, traukulius, kepenų ir blužnies padidėjimą, veido bruožų šiurkštumas, skeleto nelygumai, sąnarių sustingimas, pilvo pūtimas, raumenų silpnumas, perdėta reakcija į nuostabą ir problemos su eisena. Maždaug pusei sergančiųjų akyje atsiranda vyšnių raudonumo dėmės. Vaikai gali būti kurčias ir akli iki 1 metų ir dažnai miršta iki 3 metų nuo širdies komplikacijų arba plaučių uždegimas.
- Vėlyvas vaikas GM1 gangliozidozė dažniausiai prasideda nuo 1 iki 3 metų amžiaus. Neurologiniai simptomai yra ataksija, traukuliai, demencija ir sunku kalbėti.
- GM1 gangliozidozė išsivysto nuo 3 iki 30 metų amžiaus. Simptomai yra sumažėjusi raumenų masė (raumenų atrofija), neurologinės komplikacijos, kurios yra ne tokios sunkios ir progresuoja lėčiau nei kitos formos. sutrikimai, kai kurių žmonių ragenos drumstumas ir nuolatiniai raumenų susitraukimai, sukeliantys sukimąsi ir pasikartojančius judesius arba nenormalias laikysenas (distonija). Angiokeratomos gali išsivystyti apatinėje kūno dalyje. Daugumos paveiktų žmonių kepenų ir blužnies dydis yra normalus.
GM2 gangliozidozė.
GM2 gangliozidozė taip pat sukelia rūgščių riebalų perteklių organizme kaupiasi audiniuose ir ląstelėse, pirmiausia nervų ląstelėse. Šie sutrikimai atsiranda dėl fermento beta-heksosaminidazės trūkumo. GM2 sutrikimai apima:
- Tay-Sachs liga (taip pat žinomas kaip GM2 gangliozidozės B variantas) ir jo variantų formas sukelia fermento heksosaminidazės A trūkumas. Sergantys vaikai normaliai vystosi per pirmuosius kelis gyvenimo mėnesius. Simptomai prasideda nuo 6 mėnesių amžiaus ir apima laipsnišką protinio pajėgumo praradimą, demenciją, susilpnėjusį akių kontaktą, padidėjusį stulbinantis atsakas į triukšmą, progresuojantis klausos praradimas, sukeliantis kurtumą, rijimo pasunkėjimas, aklumas, vyšnių raudonumo dėmės ant tinklainės ir kai kurios paralyžius. Priepuoliai gali prasidėti antraisiais vaiko gyvenimo metais. Kūdikiai ilgainiui gali tapti priklausomi nuo maitinimo vamzdelio ir dažnai miršta sulaukę 4 metų nuo pasikartojančių infekcijų. Deja, specialaus gydymo nėra. Prieštraukuliniai vaistai iš pradžių gali kontroliuoti traukulius. Kiti palaikomieji gydymo būdai apima tinkamą mitybą ir hidrataciją bei kvėpavimo takų atvėrimo metodus. Reta sutrikimo forma vadinama vėlyva Tay-Sachs liga, pasireiškia žmonėms nuo 20 iki 30 metų ir jam būdinga nestabili eisena ir progresuojantis neurologinis pablogėjimas.
- Sandhoffo liga (AB variantas) yra sunki Tay-Sachs ligos forma. Paprastai jis pasireiškia sulaukus 6 mėnesių amžiaus ir neapsiriboja jokia etnine grupe. Neurologiniai požymiai gali būti laipsniškas centrinės nervų sistemos, motorikos pablogėjimas silpnumas, ankstyvas aklumas, stipri išsigandusi reakcija į garsą, spazmiškumas, šokas ar trūkčiojimas raumenys (mioklonusas), epilepsija, nenormaliai padidėjusi galva (makrocefalija) ir raudonos dėmės akyje. Kiti simptomai gali būti dažnos kvėpavimo takų infekcijos, širdies ūžesys, lėlę primenantys veido bruožai ir kepenų bei blužnies padidėjimas. Specifinio Sandhoffo ligos gydymo nėra. Kaip ir Tay-Sachs ligos atveju, palaikomoji priežiūra apima visą laiką atvirų kvėpavimo takų palaikymą, taip pat tinkamą mitybą ir hidrataciją. Prieštraukuliniai vaistai iš pradžių gali kontroliuoti epilepsiją (priepuolius).
Krabbe liga(taip pat žinomas kaip rutulinių ląstelių leukodistrofija ir galaktozilceramido lipidozė) yra autosominė recesyvinė liga, kurią sukelia fermento galaktocerebrozidazės trūkumas. Liga dažniausiai suserga kūdikiams, kurie prasideda iki 6 mėnesių amžiaus, tačiau gali pasireikšti ir paauglystėje ar pilnametystėje.
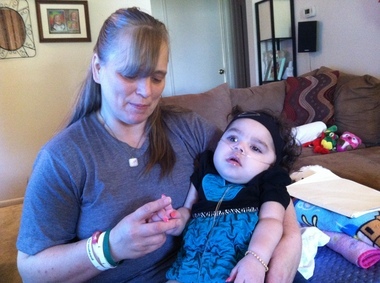
Nesuvirškintų riebalų kaupimasis neigiamai veikia nervo apsauginio apvalkalo (mielino apvalkalo) augimą ir sukelia rimtus protinių bei motorinių įgūdžių sutrikimus. Kiti simptomai yra raumenų silpnumas, sumažėjęs raumenų gebėjimas temptis (hipotonija), raumenų įtampa (spastiškumas), staigus šokas. arba galūnių trūkčiojimas (miokloniniai traukuliai), dirglumas, nepaaiškinamas karščiavimas, kurtumas, aklumas, paralyžius ir rijimo pasunkėjimas. Taip pat gali atsirasti ilgalaikis svorio kritimas.
Liga gali būti diagnozuojama ištyrus fermentus ir nustatant jai būdingą ląstelių grupę į globoidinius kūnus baltojoje smegenų medžiagoje, nervų demielinizaciją ir degeneraciją bei smegenų ląstelių sunaikinimą. Kūdikiams liga dažniausiai būna mirtina iki 2 metų amžiaus. Žmonėms, sergantiems labiau pažengusia ligos forma, ligos eiga yra švelnesnė, jie gyvena žymiai ilgiau.
Specifinis Krabbe ligos gydymas nebuvo sukurtas, nors ankstyva kaulų čiulpų transplantacija gali padėti kai kuriems žmonėms. Žmonėms, sergantiems labiau pažengusia ligos forma, ligos eiga yra švelnesnė, jie gyvena žymiai ilgiau.
Metachromatinė leukodistrofija, arba MLD, yra sutrikimų grupė, kuriai būdingas riebalinių medžiagų kaupimasis baltojoje centrinės nervų sistemos medžiagoje ir periferiniuose nervuose bei tam tikru mastu inkstuose. Panašiai kaip Krabbe liga, MLD veikia mieliną, kuris dengia ir apsaugo nervus. Šį autosominį recesyvinį sutrikimą sukelia fermento arilsulfatazės A trūkumas. Šis sutrikimas pasireiškia tiek vyrams, tiek moterims.
Metachromatinė leukodistrofija turi tris būdingas formas: vėlyvą kūdikį, nepilnametį ir suaugusiųjų.
- Vėlyvas kūdikių MLD paprastai prasideda nuo 12 iki 20 mėnesių po gimimo. Vaikai iš pradžių gali atrodyti normalūs, tačiau jiems sunku vaikščioti, jie linkę kristi, kartu su periodišku rankų ir kojų skausmu. laipsniškas regėjimo praradimas, dėl kurio atsiranda aklumas, vystymosi vėlavimas ir anksčiau įgytų etapų praradimas, rijimo sutrikimas, traukuliai ir demencija iki 2 metų. Vaikams taip pat išsivysto laipsniškas raumenų nykimas ir silpnumas ir galiausiai prarandamas gebėjimas vaikščioti. Dauguma vaikų, sergančių šia sutrikimo forma, miršta iki 5 metų amžiaus.
- Nepilnamečių MLD dažniausiai prasideda nuo 3 iki 10 metų amžiaus. Simptomai yra sutrikęs mokyklos darbas, psichikos sutrikimas, ataksija, traukuliai ir demencija. Simptomai progresuoja, mirtis įvyksta praėjus 10–20 metų nuo ligos pradžios.
- Suaugusiųjų forma. Simptomai suaugusieji prasidėti sulaukus 16 metų ir gali apimti ataksiją, traukulius, nenormalų galūnių tremorą (tremorą), susilpnėjusią koncentraciją, depresija, psichikos sutrikimai ir demencija. Paprastai mirtis įvyksta praėjus 6–14 metų nuo simptomų atsiradimo.
MLD negalima išgydyti. Gydymas yra simptominis ir palaikomasis. Kai kuriais atvejais kaulų čiulpų transplantacija gali sulėtinti ligos progresavimą. Didelė pažanga padaryta naudojant genų terapiją gyvūnų modeliuose su MLD ir klinikiniuose tyrimuose.
Wolmano ligataip pat žinomas kaip lizosomų rūgšties lipazės trūkumasyra rimtas lipidų kaupimosi sutrikimas, kuris paprastai būna mirtinas sulaukus 1 metų. Šiam autosominiam recesyviniam sutrikimui būdingas cholesterolio esterių (dažniausiai cholesterolio transportavimo formos) kaupimasis ir trigliceridai (cheminė forma, kuria organizme egzistuoja riebalai), kurie gali labai susikaupti ir pakenkti ląstelėms ir audiniai.
Tiek vyrai, tiek moterys kenčia nuo šio sutrikimo. Gimę kūdikiai yra normalūs ir aktyvūs, tačiau greitai išsivysto progresuojantis psichikos sutrikimas, padidėja kepenys ir labai padidėja blužnis, atsiranda pilvo pūtimas, virškinimo trakto problemos. gelta, anemija, vėmimas ir kalcio nuosėdos antinksčių liaukostodėl jie sukietėja.
Kitas tipas Lizosomų rūgšties lipazės trūkumas yra cholesterolio esterio kaupimosi liga. Ši itin reta liga atsiranda dėl cholesterolio esterių kaupimosi ir trigliceridai kraujo ląstelėse, limfinis ir limfoidinis audinys. Suaugusiesiems vaikams padidėja kepenys, dėl to išsivysto cirozė ir lėtinis kepenų nepakankamumas. Vaikams taip pat gali būti kalcio nuosėdų antinksčiuose, o vėlesnėse ligos stadijose gali išsivystyti gelta.
Šiuo metu fermentų pakeitimo klausimas aktyviai tiriamas tiek sergant Wolmano liga, tiek dėl cholesterolio esterio kaupimosi.
Dėmesio! Informacija skirta tik informaciniams tikslams ir nėra skirta diagnozuoti ir skirti gydymą. Visada pasitarkite su gydytoju specialistu!


